
Ce billet fait suite à ce tweet dans lequel était posé fort justement la question de la pertinence d’un traitement par bêta-bloquant (BB) en cas de cardiothyréose.
Je vais donc reprendre ici une partie de ma présentation faite lors des Journées Européennes de la Société Française de Cardiologie, en janvier 2019, en l’étoffant un peu, puisque je n’ai pas de contrainte de temps. Ce billet n’abordera pas la question de l’amiodarone que je traiterai dans un billet dédié. Je reviendrai également dans le futur sur le lien entre hypothyroïdie et cœur et en particulier insuffisance cardiaque (IC), sujet ouvrant d’intéressantes questions thérapeutiques…
Quelques définitions pour commencer
L’hyperthyroïdie est une anomalie biologique secondaire à l’hyperfonctionnement de la glande thyroïde, caractérisée par une baisse de la TSH et une élévation de la thyroxine (T4) et de la triiodothyronine T3. L’hyperthyroïdie fruste est une baisse isolée de la TSH, en l’absence d’élévation des hormones périphériques. Et on dit bien « fruste » (atténué, grossier) et non pas « frustre », qui n’existe pas mais serait dérivé de frustré (qui ressent de la frustration).
La thyrotoxicose est l’expression clinique de l’hyperthyroïdie. En cas d’implication cardiaque lors d’une thyrotoxicose, on parle de cardiothyréose.
Un peu d’épidémiologie : l’hyperthyroïdie, c’est grave docteur ?
Dans une étude de cohorte portant sur 5.599 patients, Chen et al ont montré qu’une hyperthyroïdie (définie par une TSH inférieure à 0,45 mUI/L) (ce qui ne concernait que 3% de la cohorte) était associée à un surcroit de mortalité, et dans un autre étude de cohorte portant sur plus de 25.000 patients, une TSH inférieure à 0,10 mUI/L était associée à un risque accru d’IC. Plus spécifiquement dans l’IC, dans une analyse ancillaire de l’étude SCD-HF (je reviendrai en détail sur SCD-HF quand je parlerai de l’amiodarone), la présence d’une hyperthyroïdie (définie par une TSH inférieure à 0,3 mUI/l) était associée à une risque accru de mortalité totale de 85 % (HR 1,85 ; 95 CI 1,21 – 2,83). Au passage, dans ces études, l’hypothyroïdie est au moins aussi délétère que l’hyperthyroïdie.
Toutes ces études ont pour principale limite de ne donner de corrélation qu’avec les valeurs de TSH, sans savoir s’il s’agit hyperthyroïdie fruste ou franche, la T3 et la T4 n’étant jamais connues (nous verrons dans un prochain billet concernant l’hypothyroïdie que l’ignorance des valeurs de T3 pose un problème pour identifier une forme particulière de dysfonction thyroïdienne). Et il n’y a, en dépit de ces données et des recommandations des sociétés savantes d’endocrinologie, à ce jour (à ma connaissance en tout cas), aucune étude validant la prise en charge thérapeutique d’une hyperthyroïdie fruste.
Petits rappels sur la physiologie cardiomyocytaire
Pour bien comprendre ce qui va suivre, je suis obligé de revenir sur quelques bases de physiologie cardiaque. Comment se fait la contraction et la relaxation d’un cardiomyocyte ?
Cycle calcique
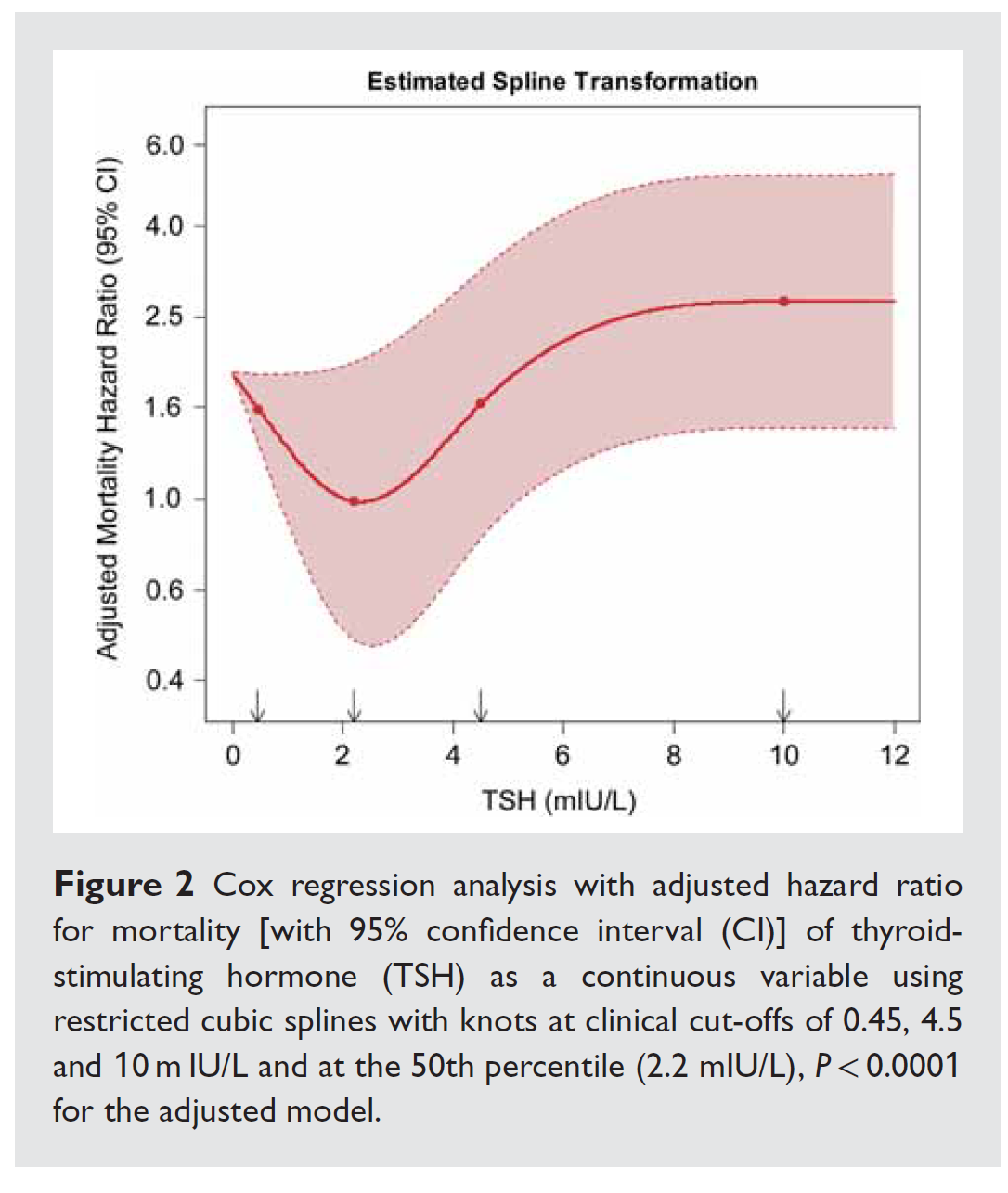
Un élément majeur impliqué dans la contraction est le calcium. Il est stocké en grandes quantités dans le réticulum sarcoplasmique. L’arrivée d’un potentiel d’action va ouvrir des canaux calciques L membranaires, permettant l’entrée de petites quantités de calcium dans le cytosol. Ce calcium va déclencher la sortie du calcium sarcoplasmique, via les récepteurs à la ryanodine (RyR), ce qui va permettre la contraction. Lors de la relaxation, le calcium est recapté dans le réticulum sarcoplasmique par la sarcoendoplasmic reticulum Ca2+-ATPase (SERCA) (ce qui me permet de rappeler que la relaxation du muscle cardiaque est un processus qui consomme de l’énergie…).
D’après Braunwald Heart Disease : a textbook
SERCA est elle-même régulée par le phospholamban. Le phospholamban, à l’état normal, inhibe SERCA, et empêche donc la recapture du calcium. Quand le phospholamban est phosphorylé, il ne joue plus son rôle, et la recapture du calcium vers le réticulum, via SERCA, est facilitée, ce qui améliore la relaxation cardiomyocytaire.
Une petite partie du calcium est également évacuée du cytosol dans l’espace intercellulaire par l’échangeur Sodium/Calcium qui va faire entrer du sodium dans la cellule en suivant le gradient ionique, en échange d’une sortie de calcium. Ce flux n’est possible que si le gradient de sodium entre le cytosol et le milieu extracellulaire est suffisant. La création de ce gradient est facilité l’action de la Na/K-ATPase qui fait sortir de la cellule le sodium en échange de potassium.
La signalisation béta-adrénergique

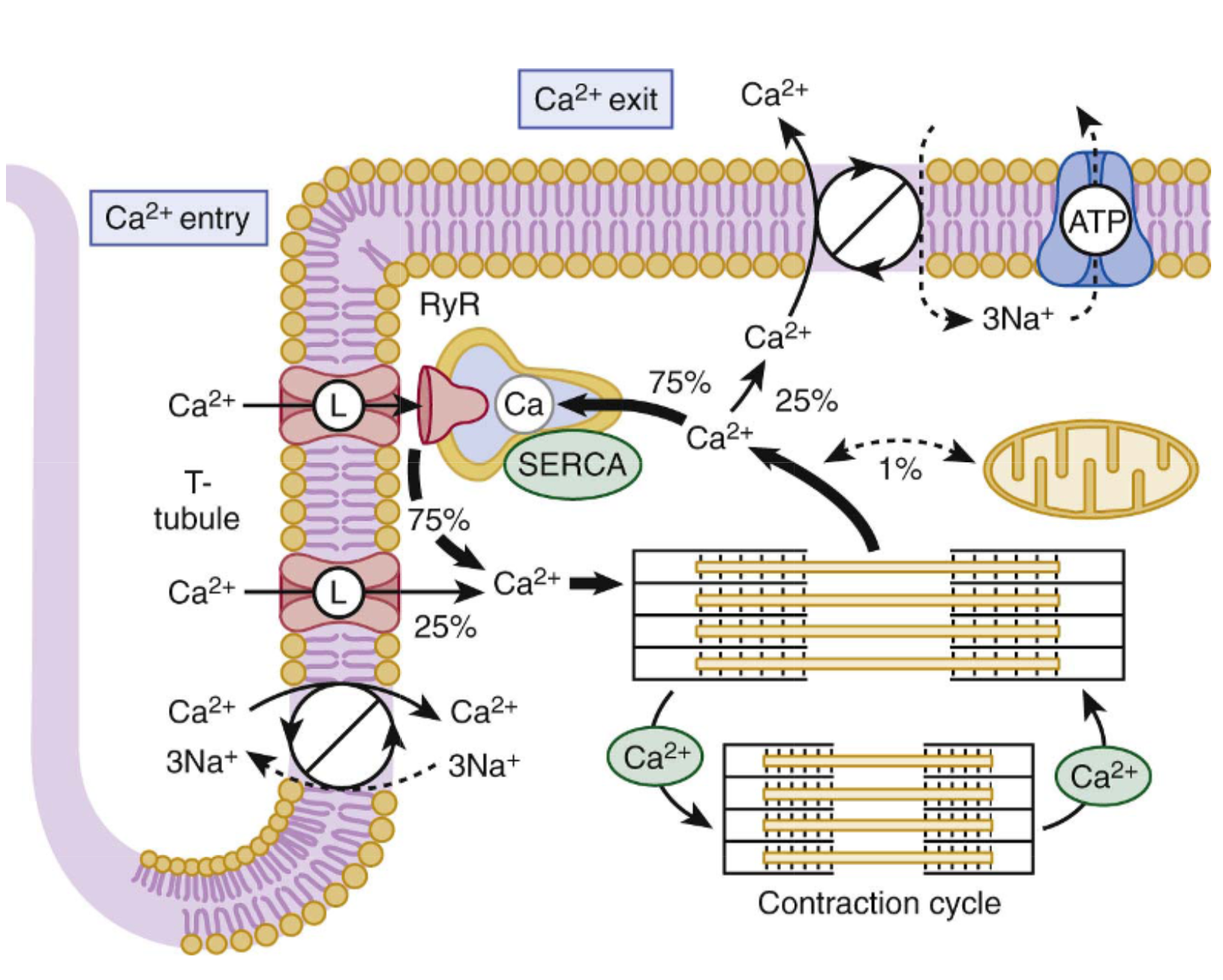
D’après Braunwald Heart Disease : a textbook
Les récepteurs béta-adrénergiques sont des récepteurs à 7 domaines transmembranaires couplés aux protéines G. La stimulation du récepteur entraine l’activation de la protéine Gs qui régule différents effecteurs, comme l’adenylate cyclase ou les canaux ioniques notamment calciques L dont j’ai parlé plus haut.
L’activation de l’adenylate-cyclase puis de la protéine kinase A (PKA) va favoriser l’ouverture des canaux calciques et de RyR, et l’entrée de calcium dans le cytosol, facilitant l’augmentation du calcium intracellulaire. Ceci favorise la contraction cardiaque (effet inotrope positif). L’action sur les canaux calciques (via l’adenylate-cyclase ou directement par la protéine Gs) favorise également la dépolarisation cardiomyocytaire et entraine une accélération du rythme cardiaque (effet chronotrope positif). Pour être complet, la PKA va également phosphoryler le phospholamban, ce qui le rend inactif ; cette inactivation empêche l’inhibition de SERCA et entraine une meilleure recapture du calcium, améliorant la relaxation ventriculaire (effet lusitrope positif).
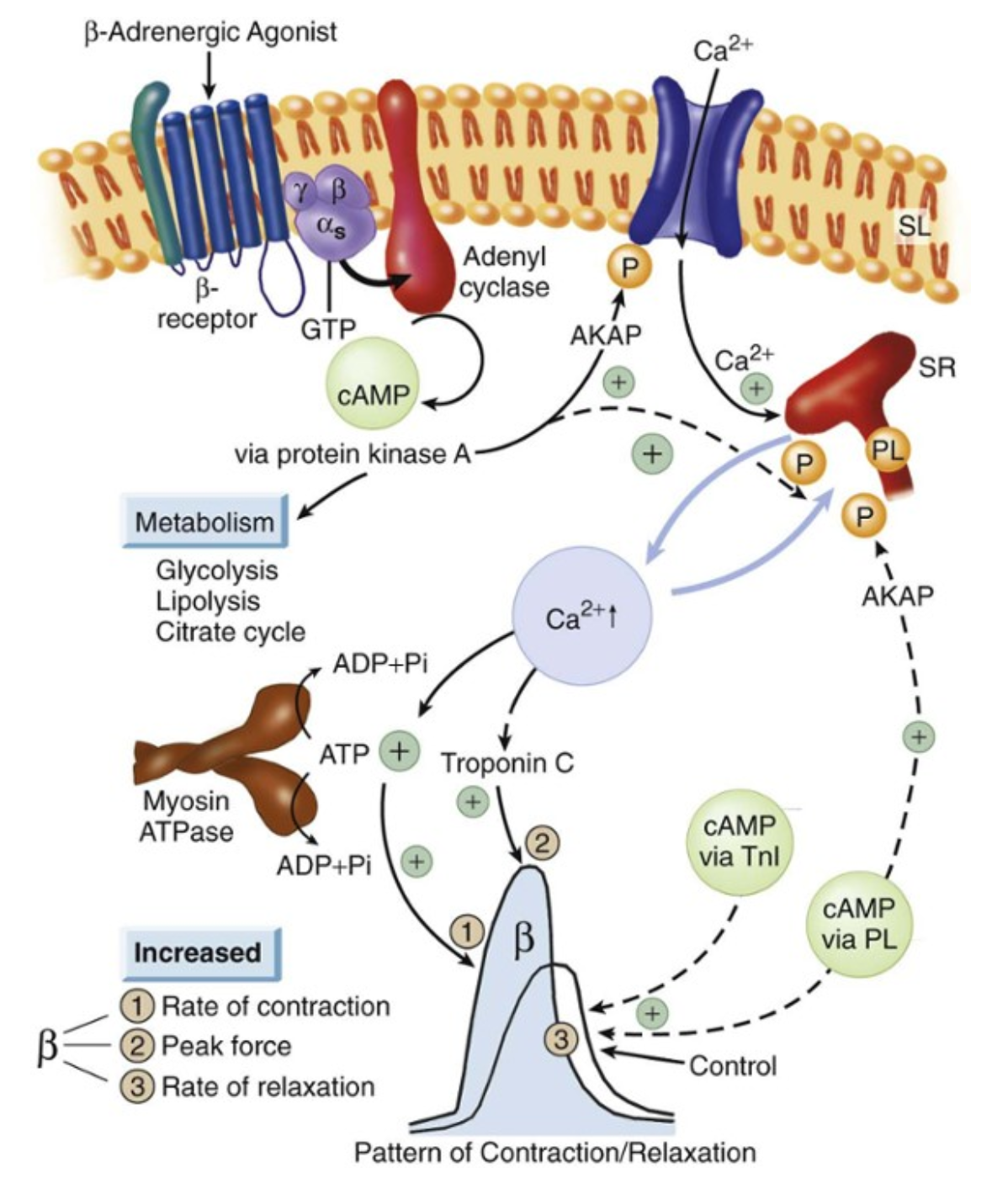
D’après Braunwald Heart Disease : a textbook
Conséquences cardiovasculaires de l’hyperthyroïdie : la cardiothyréose
Effets cardiomyocytaires des hormones thyroïdiennes
La thyroïde sécrète, sous l’influence de la TSH hypophysaire, de la T4 (à 85%) et de la T3, hormone responsable de l’activité biologique. La T4 est convertie en T3 au niveau du foie, des reins et des muscles squelettiques.
Les cardiomyocytes possèdent des récepteurs à la T3, mais sont incapables de transformer la T4 en T3, et ne possèdent pas de récepteur à la T4. Ils sont donc totalement dépendants du taux circulant de T3. Une fois dans le cardiomyocytes, la T3 exerce des effets génomiques et non-génomiques sur la cellule. Ces effets transcriptionnels et non transcriptionnels agissent de concert pour majorer les fonctions systolique et diastolique, favorisant à la fois l’augmentation de la capacité contractile mais aussi du remplissage ventriculaires gauches.
Effets de la T3
Une fois entrée dans le noyau, la T3 se fixe sur un récepteur nucléaire (TR), qui va induire la transcription en se fixant sur la région promotrice des gènes régulés positivement par l’intermédiaire d’un TRE (Thyroid hormone Response Element). La fixation du TRE lié au couple T3-TR permet l’activation de la transcription, alors qu’en l’absence de T3, la fixation du TRE entraine l’inactivation de la transcription de ces mêmes gênes. Par ce biais, la T3 stimule par exemple la synthèse des chaines alpha de la myosine (myosine rapide à activité ATPase élevée) ou de SERCA.
Le TRE exerce également une régulation négative sur certains gènes, c’est à dire que leur transcription est inhibée par la fixation du TRE lié au couple T3-TR. C’est notamment le cas des gènes de la chaine bêta de la myosine ou du phospholamban.
Le tableau ci-dessous reprend certaines gènes modulés par l’action de la T3.
| Régulation positive (promotion par la T3) | Régulation négative (inhibition par la T3) |
| Chaine alpha de la myosine | Chaine bêta de la myosine |
| SERCA | Phospholamban |
| Na/K-ATPase | Echangeur Sodium/Calcium |
| Récepteurs bêta-adrénergiques | Adenylate-cyclase |
| Protéines G |
La T3 a des effet extranucléaires non-génomiques sur les cardiomyocytes, ainsi que sur la musculature vasculaire. Ces effets apparaissent rapidement, ne dépendant pas d’un processus transcriptionnel. Il s’agit notamment de modifications de divers canaux ioniques membranaires ou de protéines mitochondriales.
Au niveau des muscles lisses, la T3 stimule (par voies génomique et non génomique) la NO synthase, et agit également par le biais des mêmes modifications du cycle calcique, facilitant la relaxation musculaire lisse.
Conséquences des effets génomiques et non-génomiques de la T3
La baisse de la synthèse du phospholamban diminue l’inhibition pesant sur SERCA, dont l’expression est elle-même augmentée. Cette régulation opposée favorise la recapture du calcium intracellulaire vers le réticulum endoplasmique, ce qui a pour effet d’accélérer et de favoriser la relaxation cardiomyocytaire.
D’après Klein et al ; NEJM; 2001
Sous l’effet de l’augmentation de la T3, on observe une augmentation du nombre de récepteurs bêta-adrénergiques, responsable d’une augmentation de la sensibilité à la stimulation adrénergique. Bien qu’il y ait une augmentation des bêta-récepteurs et des protéines Gs, la baisse de l’expression de l’adenylal cyclase entraine un découplage entre la stimulation adrénergique et la réponse contractile, en dépit d’une sensibilité adrénergique globale normale voire augmentée. L’effet inotrope de la T3 est donc indépendant de l’axe adrénergique, et passe par la promotion des chaines alpha de la myosine et par l’augmentation de la concentration du réticulum sarcoplasmique en calcium du fait de l’hyperactivité de SERCA. En revanche, l’absence d’inhibition de la protéine Gs préserve l’action des bêta-récepteurs sur les canaux calciques L, et, de ce fait, sur la fréquence cardiaque (FC), et dans une bien moindre mesure sur l’inotropisme
Traductions cliniques
La cardiothyréose, expression cardiaque de la thyrotoxicose, peut se manifester par des palpitations, une tachycardie, une intolérance à l’exercice, une dyspnée d’effort, un élargissement de la différentielle tensionnelle et hypertension artérielle systolique, et parfois une fibrillation atriale. Bien que l’état cardiaque ressemble à un état d’hyperadrénergie, le taux de catécholamines n’est pas augmenté, voire est même diminué.
Cardiopathie thyréotoxique
La baisse des résistances périphériques entraine une baisse de la pression artérielle moyenne, responsable d’une baisse de la postcharge ventriculaire gauche, favorisant l’activation du système rénine-angiotensine-aldostérone (SRAA) et donc la réabsorption hydrosodée. En parallèle la T3 favorise l’érythropoïèse et stimule également la synthèse de rénine (voir ici un rappel succinct du fonctionnement du SRAA). L’ensemble de ces phénomènes conduisent à une augmentation du volume sanguin et de la précharge ventriculaire gauche.
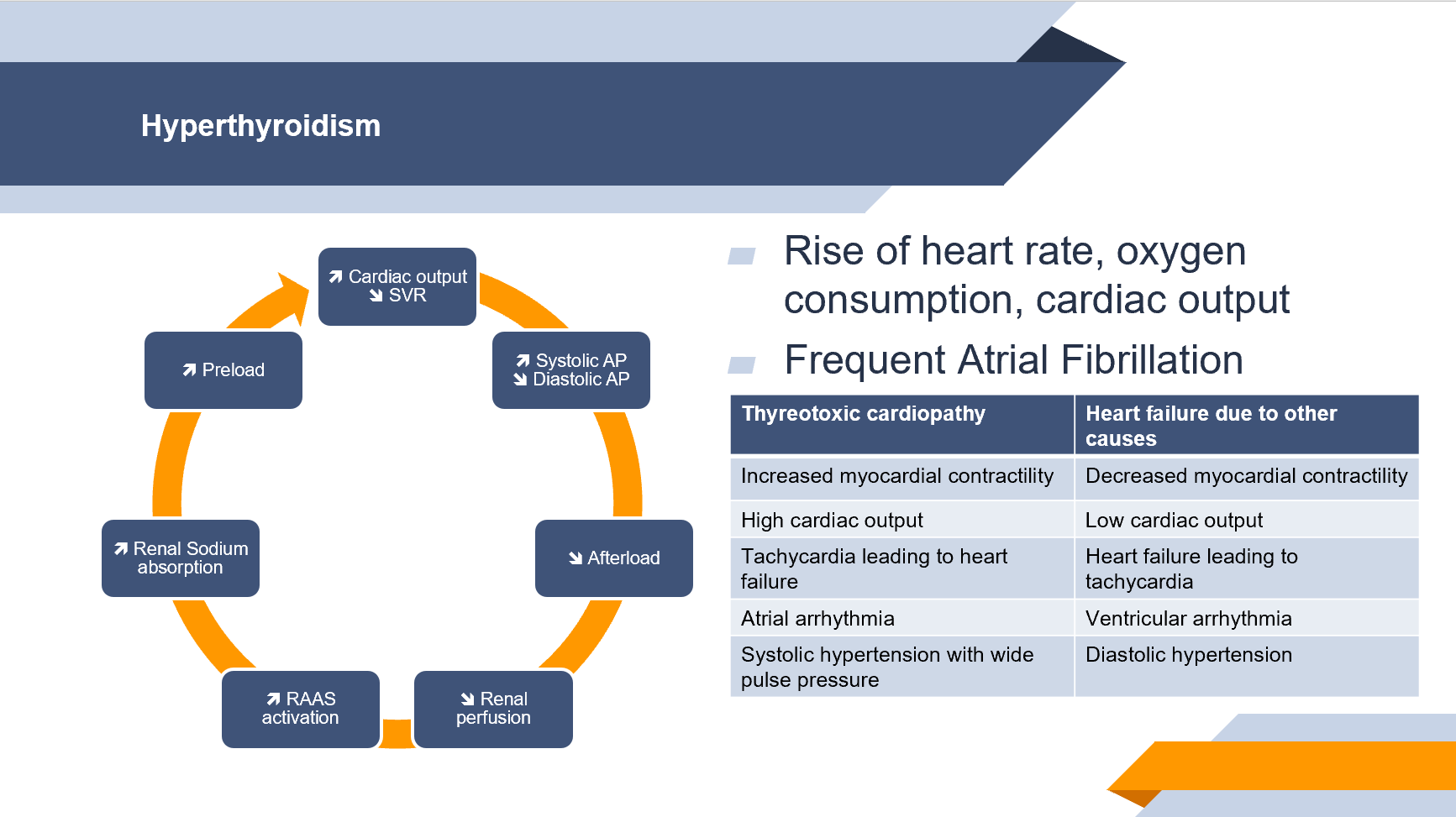
L’augmentation du débit cardiaque peut être très importante, pouvant atteindre 50 à 300 % du débit habituel, effet conjoint de l’élévation de la FC, de l’amélioration de l’inotropisme et du lusitropisme, de l’augmentation du volume sanguin et de la baisse des résistances périphériques. Cet état d’hyperdébit est responsable d’une intolérance à l’effort liée à la tachycardie et la vasodilatation de repos, rendant moindre les possibilité d’adaptation à l’effort. On ne peut donc pas à proprement parler d’une IC, car la capacité du cœur à préserver un débit au repos et à l’effort est en fait préservée. On peut parler néanmoins de cardiopathie thyréotoxique. La prolongation de l’hyperthyroïdie mène avec le temps à l’installation d’une hypertrophie ventriculaire gauche, puis à une dilatation et une altération de la FEVG, avec authentique IC. On pourrait décrire 3 stades à la cardiomyopathie thyréotoxique :
- Phase hyperkinétique : fonctions ventriculaires gauches préservées mais altération de l’adaptabilité à l’effort du fait de l’hyperdynamie de base.
- Phase nomokinétique : étape compensatoire avec hypertrophie ventriculaire gauche réversible et débit cardiaque conservé.
- Phase hypokinétique : stade décompensé avec baisse du débit cardiaque et du volume d’éjection systolique, hypertrophie et dilatation ventriculaire gauches. Authentique IC
Une part des altérations de la contractilité cardiaque et du débit pourrait aussi se rapprocher des tableaux de cardiopathies rythmiques, du fait de la tachycardie incessante. L’existence d’une cardiopathie sous-jacente peut également rendre difficile l’adaptation à l’augmentation de la demande métabolique liée à l’hyperthyroïdie, en particulier en cas de passage en fibrillation atriale.
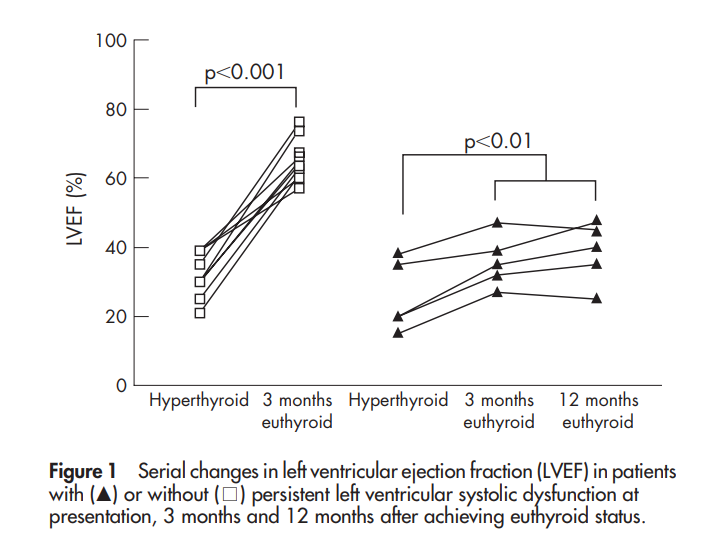
D’après Siu et al ; Heart ; 2007
Sur une série de 590 patients consécutifs, Siu et al retrouvaient une incidence d’IC (définie selon les critères de Framingham) de 5,8 % (34 patients). Ces patients étaient plus souvent de sexe masculin, plus âgés et avec plus de comorbidités cardiovasculaires et pour 94 % d’entre eux en fibrillation atriale (FA). L’existence d’une FA au moment du diagnostic est une facteur fort d’apparition d’une IC (OR 37,4 ; 95% CI 9,7 – 144,0). Seulement la moitié environ des patients avec signes et symptômes d’IC avait une baisse de la FEVG. Quelques patients, dont la FEVG ne s’était pas corrigée après 3 mois de traitement, n’ont pas normalisé leur FEVG, sans que ne soit identifié de facteur prédisposant à une récupération. Cela concerne 5 patients, soit 15 % des 34 avec signes et symptômes d’IC et 31 % sur les 16 ayant une altération de la FEVG. Au final, une altération de FEVG sous forme de cardiopathie dilatée persiste chez environ 1% des patients ayant une hyperthyroïdie. Un patient est décédé d’IC réfractaire.
Fibrillation atriale
La FA est fréquente en cas d’hyperthyroïdie, touchant, selon les études et les cohortes, de 2 à 20 % des patients, avec un risque similaire que l’hyperthyroïdie soit fruste ou franche (voir par exemple ici). Bien que l’hyperthyroïdie ne soit finalement responsable que d’environ 1% des FA, l’analyse du bilan thyroïdien est indispensable devant toute FA. La FA est probablement la conséquence des modifications d’expression des canaux ioniques, responsables d’un raccourcissement du potentiel d’action, notamment au niveau atrial, favorisant les circuits de ré-entrée. Le risque de FA augmente avec l’âge et la présence de comorbidités.
Notons qu’on observe de manière homogène dans les publications, dès l’hyperthyroïdie fruste, une accélération de la FC et une augmentation de la charge en extrasystoles atriales et ventriculaires (voir par exemple ici). Les résultats sur la fonction cardiaque en cas d’hyperthyroïdie fruste sont plus discordants, probablement en raison de cohortes de caractéristiques différentes en terme d’âge, de niveau de TSH et de durée d’évolution.
Dans la publication de Siu et al, parmi les patients ayant des signes d’IC (les données ne sont pas citées pour les patients n’ayant pas d’IC), le retour spontané en rythme sinusal se fait chez 40 % des patients, en moyenne au bout de 121 (+/- 46) jours pour les patients sans altération de la FEVG, et 328 (+/- 131) jours en cas de baisse de la FEVG.
Autres atteintes cardiovasculaires
40 % des patients ayant une cardiothyréose présente une hypertension pulmonaire, du fait de l’hyperdébit, de l’éventuelle dysfonction ventriculaire gauche ou, peut-être, d’une authentique atteinte vasculaire avec prolifération cellulaire.
L’augmentation de la demande en oxygène liée à l’hyperthyroïdie peut entrainer des anomalies de la réserve coronaire et une ischémie myocardique, notamment chez le sujet âgé, sans qu’il n’existe forcement de lésions des gros troncs coronaires.
Prise en charge thérapeutique
L’inhibition de l’hyperactivation thyroïdienne est évidement primordiale pour « rafraichir » le patient. Les BB ont un effet rapide permettant de juguler les effets périphériques et d’attendre les effets très décalés des thérapies antithyroïdiennes (antithyroïdiens de synthèse, IRAthérapie, chirurgie). En cas de cardiothyréose, qui peut entrainer des complications graves en particulier sur un terrain d’IC sous jacent, l’usage de glucorticoides ou de perchlorate de potassium peut être nécessaire afin d’accélérer la baisse de la sécrétion de T4 et de T3. Les inhibiteur calciques doivent être évités en raison des effets sur le muscle lisse et le risque de baisse de pression artérielle, et les digitaliques sont en général peu efficaces.
L’adjonction d’un traitement diurétique est tout à fait possible si le contexte clinique le justifie. Si la pression artérielle le permet, un traitement par inhibiteur de l’enzyme de conversion peut permettre d’améliorer le contrôle de l’hyperactivation du SRAA, ce d’autant que la stimulation de la rénine par la T3 aggrave encore le tableau.
Les bêta-bloquants
Les BB ont une place centrale dans la prise en charge de la cardiothyréose, tant qu’une altération de la FEVG ne s’est pas installée. En effet, ils permettent de diminuer le débit cardiaque et donc la consommation en oxygène du myocarde, en corrigeant la part rythmique de la cardiopathie, sans avoir d’effet délétère sur l’inotropisme.
Rappelons en effet les mécanismes physiopathologiques de la cardiopathie thyréotoxique. La stimulation par la T3 augmente le nombre de bêta-récepteur et donc la sensibilité catécholergique. Néanmoins, si les bêta-récepteurs reste couplés aux canaux calciques L via la protéine Gs, la baisse de la synthèse de l’adenylal-cyclase sous l’effet de la T3 découple en grande partie les bêta-récepteurs du cycle calcique. Les BB diminuent donc l’ouverture des canaux calciques L, ce qui ralenti la dépolarisation cardiomyocytaire, mais n’a pas d’action sur l’inotropisme.

d’après Mintz et al ; JCEM ; 1991
Ce concept a été illustré dans une petite étude prospective, non randomisée, au cours de laquelle 9 femmes en hyperthyroïdie ont bénéficié d’un traitement par 160 mg de PROPRANOLOL, en plus du traitement antithyroïdien. Le groupe contrôle comprenait 10 patientes euthyroidiennes appariées selon l’âge. Le PROPRANOLOL permettait un ralentissement significatif de la FC passant de 97 +/- 15 bpm à 80 +/- 12 bpm, sans modification de la FEVG ou du temps de relaxation isovolumique ventriculaire gauche (une estimation de la fonction diastolique). L’amélioration de la fonction diastolique n’intervient que lors du retour en euthyroïdie.
Le PROPRANOLOL est classiquement utilisé dans cette indication en raison de son caractère non sélectif pour les récepteurs bêta-1 ou bêta-2. Ces derniers sont présents sur les muscles lisses, ce qui permet de lutter contre la vasodilation périphérique et la baisse des résistances vasculaires. Ils sont également présents au niveau cardiaque, préférentiellement sur les cardiomyocytes atriaux. On considère habituellement que le PROPRANOLOL agit également en diminuant la conversion périphérique de T4 en T3, mais la baisse de T3 n’est probablement pas suffisante pour avoir un effet cliniquement significatif (voir notamment ici). La dose quotidienne habituellement recommandée est au moins de 160 mg (voir ici).
Évidemment, l’usage des bêta-bloquants dans la cardiopathie thyréotoxique n’est recommandée qu’en l’absence de dégradation du débit cardiaque. Plusieurs cas cliniques ont été publiés pour relater des épisodes d’arrêt circulatoire après administration de PROPRANOLOL, à chaque fois chez des patients ayant une dysfonction ventriculaire sous-jacente avec altération du débit cardiaque (voir ce papier par exemple). Une étude clinique de qualité serait évidement salutaire, mais ne sera probablement jamais réalisée…
Les digitaliques
Les digitaliques sont moins efficace en cas d’hyperthyroïdie que chez le patient euthyroïdien.
On sait depuis longtemps que l’hyperthyroïdie entraine une baisse de la concentration plasmatique des digitaliques (depuis E. Braunwald – déjà – en 1961), bien que le mécanisme reste obscur ; peut-être du fait de changements dans le volume de distribution.
L’action de la digoxine en dehors du cardiomyocyte est mal comprise, mais on sait qu’elle agit par un effet parasympathomimétique notamment au niveau du tissus atrial et du nœud atrioventriculaire. Elle diminue également, dans une moindre mesure, l’activité adrénergique, ce qui n’est, dans le cadre de l’hyperthyroïdie, que de peu d’intérêt, l’activité catécholergique n’étant pas augmentée.
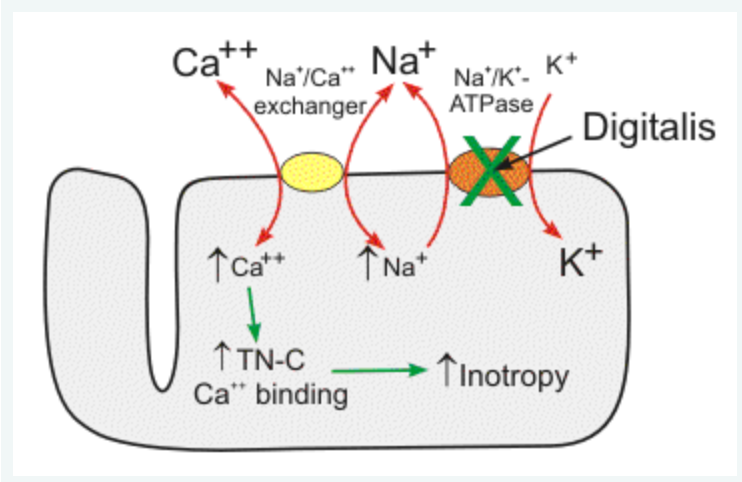
Au niveau cardiomyocytaire, la digoxine agit sur la Na/K-ATPase, qu’elle inhibe. Elle empêche donc la sortie de sodium de la cellule. L’échangeur Sodium/Calcium (bidirectionnel mais faisant, la majorité du temps, entrer du sodium pour faire sortir de calcium selon les gradients de concentration), fonctionne donc moins et la concentration en calcium cytosolique augmente, ce qui explique l’effet inotrope positif de la digoxine.
En cas d’hyperthyroïdie, l’augmentation du nombre et de l’activité de la Na-K ATPase entraine une baisse du contenu intracellulaire en sodium. La baisse du contenu en sodium favorise le fonctionnement de l’échangeur Sodium/Calcium qui va donc faire entrer du sodium en échange d’une sortie de calcium. Cette baisse de la concentration calcique entraine une baisse de la réponse aux agents inotropes agissant par le biais d’une élévation du sodium intracellulaire, dont la digoxine, en raison de la baisse du flux calcique via l’échangeur (voir par exemple l’étude de Kim et al en 1984)
La fibrillation atriale
En cas de FA en contexte d’hyperthyroïdie, environ 60 % des patients retournent en rythme sinusal dans un délai de 8 à 12 semaines. La réduction spontanée a moins de chance de survenir en cas de maladie cardiaque sous-jacente, d’évolution prolongée de la cardiothyréose ou chez les patients les plus âgés.
Contrairement à ce que je dis habituellement dans le cadre de la FA « classique », une tentative de cardioversion électrique (on évitera évidement, dans ce cas, de recourir à l’amiodarone) peut se discuter, puisqu’il existe un facteur déclenchant et pérennisant évident qui aura été maitrisé.
Aucune étude n’a évalué la pertinence ou non d’une anticoagulation efficace dans la FA thyréotoxique, mais il semble licite de la proposer si les critères habituels d’anticoagulation sont remplis.
Conclusion
La thyrotoxicose est une maladie rare mais potentiellement grave qui tue par ses complications cardiaque. La cardiopathie thyréotoxique négligée va évoluer vers une forme de cardiopathie dilatée dont la récupération n’est pas certaine. Un bilan thyroïdien est donc nécessaire devant toute FA de novo et tout tableau d’hyperdébit cardiaque.
Au stade initial, la prise en charge repose sur le PROPRANOLOL qui va permettre d’améliorer l’hémodynamique en ne jouant que sur le versant fréquentiel de l’hyperdébit, sans altérer l’inotropisme. Ce traitement doit être évité ou utilisé avec précaution aux stades avancées, quand le tableau se rapproche plus d’une IC classique. Les digitaliques peuvent être utiles mais restent moins efficaces que dans les tableaux de tachycardie ou d’IC habituels.
Quelques références
J’ajoute ici les références de quelques revues de la littérature sur les relations entre cœur et thyroïde, sur lesquelles je me suis appuyé pour rédiger ce billet.
- Osuna PM, Udovcic M, Sharma MD. Hyperthyroidism and the Heart. Methodist DeBakey Cardiovasc J. 2017;13(2):60‑3.
- Vargas-Uricoechea H, Bonelo-Perdomo A. Thyroid Dysfunction and Heart Failure: Mechanisms and Associations. Curr Heart Fail Rep. 1 févr 2017;14(1):48‑58.
- Klein I, Danzi S. Thyroid Disease and the Heart. Curr Probl Cardiol. 1 févr 2016;41(2):65‑92.
- Jabbar A, Pingitore A, Pearce SHS, Zaman A, Iervasi G, Razvi S. Thyroid hormones and cardiovascular disease. Nat Rev Cardiol. janv 2017;14(1):39‑55.

Bonjour, je suis mere de deux enfants et j’utilise la teleconsultation avec mon medecin generaliste pendant la crise COVID. Ca fonctionne tres bien!
J’aimeJ’aime